DeePMD-kit
👉Вернуться к списку инструкций
![]() DeePMD-kit – это инструмент для исследования материалов на основе машинного обучения, разработанный исследовательским коллективом в Институте физики высоких температур Китайской академии наук. Этот пакет позволяет применять модели глубокого обучения для атомных систем, включая молекулярную динамику и интерпретацию свойств материалов. DeePMD-kit представляет собой решение, которое объединяет алгоритмы глубокого обучения с классическими методами физического моделирования для достижения высокой точности в предсказании свойств атомных систем.
DeePMD-kit – это инструмент для исследования материалов на основе машинного обучения, разработанный исследовательским коллективом в Институте физики высоких температур Китайской академии наук. Этот пакет позволяет применять модели глубокого обучения для атомных систем, включая молекулярную динамику и интерпретацию свойств материалов. DeePMD-kit представляет собой решение, которое объединяет алгоритмы глубокого обучения с классическими методами физического моделирования для достижения высокой точности в предсказании свойств атомных систем.
Официальный сайт: https://docs.deepmodeling.com/projects/deepmd/en/r2/
Доступные версии на суперкомпьютере НИУ ВШЭ
module load deepmd-kit/2.2.10 # DeePMD-kit версии 2.2.10
Обновление версий производится по запросу пользователей.
Тестовые исходные данные расположены на суперкомпьютере в каталоге
Пользователи: Лаборатория вычислительной физики, Департамент прикладной математики МИЭМ, любые подразделения НИУ ВШЭ.
Ключевые особенности
- интерфейс с TensorFlow, что делает процесс обучения автоматизированным и эффективным,
- интерфейс с пакетами классической молекулярной динамики и квантовой молекулярной динамики, включая LAMMPS, i-PI, AMBER, CP2K, GROMACS, OpenMM и ABUCUS,
- реализация моделей серии Deep Potential, которые успешно применялись к конечным и расширенным системам, включая органические молекулы, металлы, полупроводники, диэлектрики и т.д.,
- поддержка MPI и GPU, что делает его высокоэффективным для параллельных и распределенных вычислений высокой производительности,
- высокая модульность, что облегчает адаптацию к различным дескрипторам для моделей потенциальной энергии на основе глубокого обучения.
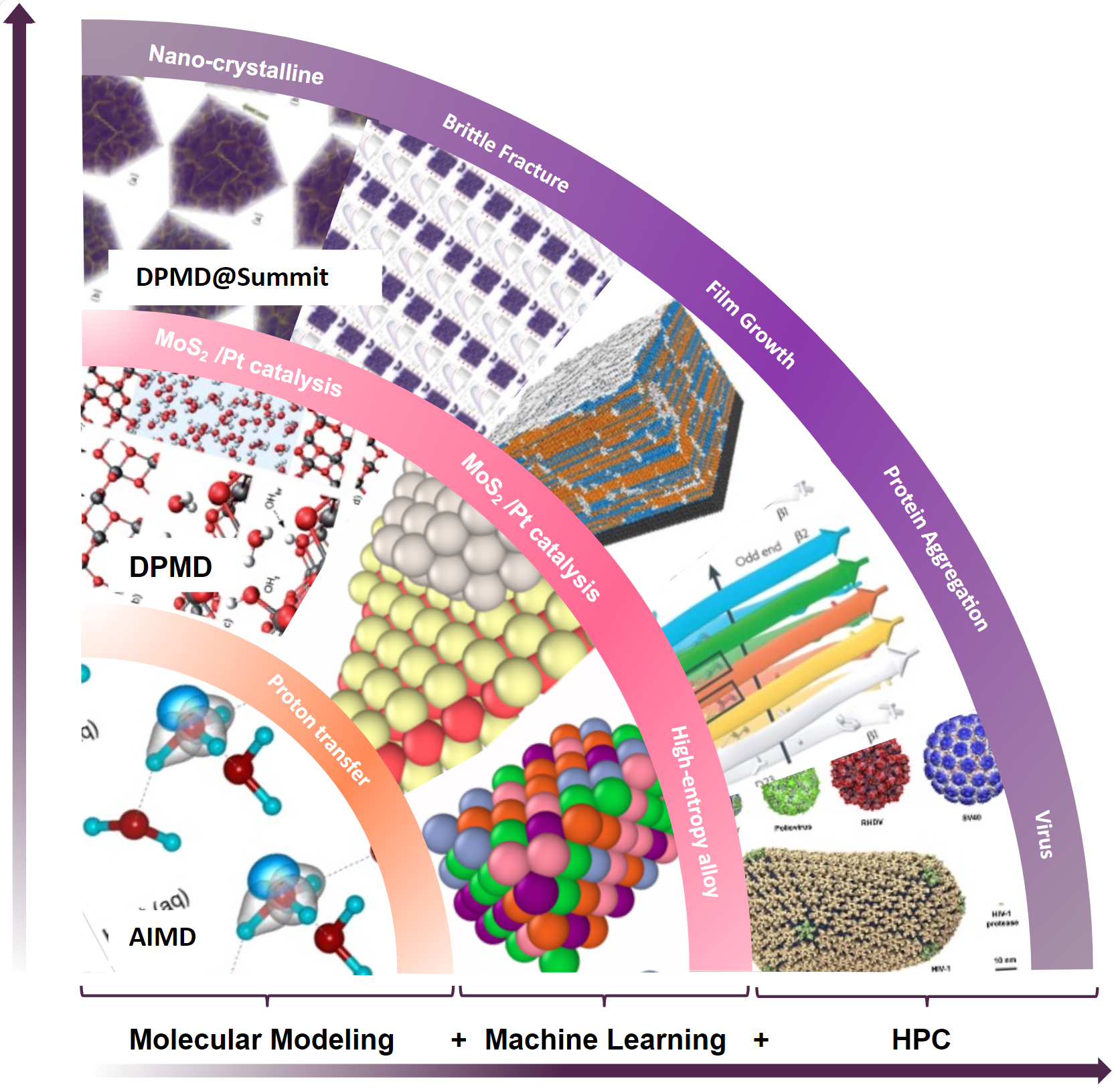
Рис. 1. Вычислительная парадигма, состоящая из молекулярного моделирования, машинного обучения и высокопроизводительных вычислений
Выполнение расчетов на суперкомпьютере
Для выполнения расчёта на суперкомпьютере необходимо подготовить скрипт-файл для очереди задач с запросом требуемых ресурсов (см. общую инструкцию по запуску задач на суперкомпьютере).
Пример запуска на GPU
В качестве примера, выполним моделирование для тестовых данных, представленных в учебном материале.
Конвертация данныхСкопируем директорию с примером в домашнюю директорию:
module load deepmd-kit/2.2.10
cd ~
cp -r /opt/software/deepmd-kit/v2/examples/CH4/ ./
Переходим в директорию с примером и обнаруживаем там файл :
cd CH4/00.data
ls
Этот файл, являющийся результатом работы пакета VASP, нужно конвертировать в формат для DeePMD-kit. Для этого запустим файл convert.py (расположен в той же директории) командой :
Содержимое файла convert.py
import dpdata
import numpy as np
data = dpdata.LabeledSystem('OUTCAR', fmt = 'vasp/outcar')
print('# the data contains %d frames' % len(data))
# random choose 40 index for validation_data
index_validation = np.random.choice(200,size=40,replace=False)
# other indexes are training_data
index_training = list(set(range(200))-set(index_validation))
data_training = data.sub_system(index_training)
data_validation = data.sub_system(index_validation)
# all training data put into directory:"training_data"
data_training.to_deepmd_npy('training_data')
# all validation data put into directory:"validation_data"
data_validation.to_deepmd_npy('validation_data')
print('# the training data contains %d frames' % len(data_training))
print('# the validation data contains %d frames' % len(data_validation))
cd ../01.train
Обучение искусственной нейронной сети
Для постановки задачи в очередь сформируем скрипт deepmd-kit.sbatch со следующим текстом:
#!/bin/bash
#SBATCH --job-name=deepmd-kit-test # Название задачи
#SBATCH --time=01:00:00 # Максимальное время выполнения (1 час)
#SBATCH --error=deepmd-kit-%j.err # Файл для вывода ошибок
#SBATCH --output=deepmd-kit-%j.log # Файл для вывода результатов #SBATCH --constraint="type_e|type_a|type_b|type_c" # Предпочитаемые типы вычислительных узлов
#SBATCH --nodes 1 # Число используемых узлов
#SBATCH --ntasks 1 # Количество MPI процессов
#SBATCH --cpus-per-task=4 # Количество CPU на MPI-процесс
#SBATCH --gpus-per-task=1 # Число используемых GPU
#SBATCH --mail-user=ваша_почта # Укажите ваш email для отправки уведомлений (если нужно)
#SBATCH --mail-type=END,FAIL # События, требующие уведомления # Деактивация окружения source deactivate
# Очистка модулей module purge # Загрузка модуля DeePMD-kit module load deepmd-kit/2.2.10 # Переменные среды, изменяющие параметры параллелизма и многопоточности для различных библиотек # Их значения подбираются экспериментально для каждой конкретной задачи export OMP_NUM_THREADS=$((SLURM_CPUS_PER_TASK / 2)) export TF_INTRA_OP_PARALLELISM_THREADS=$((SLURM_CPUS_PER_TASK / 2)) export TF_INTER_OP_PARALLELISM_THREADS=$((SLURM_CPUS_PER_TASK / 2)) # Команда запуска mpirun -l -launcher=fork -hosts=localhost -np $SLURM_GPUS dp train input.json
# Рекомендуемая пропорция ресурсов на cHARISMa: # - на узлах типов A, B, C: 1 GPU и 4 ядра CPU, # - на узлах типа E: 1 GPU и 16 ядер CPU. # Как можно больше заполняйте память GPU. Отслеживайте загрузку памяти через: https://lk.hpc.hse.ru
Для постановки задачи в очередь необходимо выполнить команду sbatch deepmd-kit.sbatch
Посмотреть состояние своих задач можно с помощью команды mj.
Результат выполнения скрипта будет сохранён в файл deepmd-kit-00000.log (где 00000 - номер задачи в очереди). Сообщения, выводимые в процессе работы DeePMD-kit, будут записаны в файл deepmd-kit-00000.err (где 00000 - номер задачи в очереди).
Сохранение модели
После обучения, нужно сохранить, а затем сжать модель (Freeze/Compress the Model):
dp freeze -o graph.pb
dp compress -i graph.pb -o graph-compress.pb
В этих командах после -o указываются имена выходных файлов, а после -i пишется название входного файла.
Тестирование качества модели
DeePMD-kit предоставляет пользователям возможность проверки качества полученной модели. Для этого нужно запустить следующую команду:
dp test -m graph-compress.pb -s ../00.data/validation_data -n 40 -d resultsЗдесь после -m указывается файл с моделью, которую необходимо проверить, после -s – путь к набору данных для тестирования, -n указывает количество проверяемых кадров, а после -d пишется имя файла для записи результатов.
Применение модели в LAMMPS
Подробнее о запуске задач LAMMPS на суперкомпьютере можно узнать в соответствующей инструкции.
Полученную модель можно использовать для симуляций молекулярной динамики. Переходим в директорию 02.lmp:
cd ../02.lmpСоздаём ссылку на модель, полученную в процессе обучения:
ln -s ../01.train/graph-compress.pbПроверяем, что в директории есть 3 файла:
ls # conf.lmp graph-compress.pb in.lammps
conf.lmpсодержит начальную конфигурацию для MD симуляции метана в газовой фазе.in.lammpsявляется скриптом для запуска LAMMPS. Этот файл является стандартным входным файлом LAMMPS для MD симуляций с двумя исключениями:
pair_style graph-compress.pb pair_coeff * *Эти строки указывают LAMMPS использовать модель Deep Potential для вычисления атомных взаимодействий, хранящуюся в файле
graph-compress.pb. Запускаем LAMMPS стандартным образом (см. инструкцию), в качестве входного файла (после флага -in) указываем in.lammps. После завершения симуляции будут сгенерированы файлы log.lammps и ch4.dump, содержащие термодинамическую информацию и траекторию молекул соответственно. Применение модели в GROMACS
Подробнее о запуске задач GROMACS на суперкомпьютере можно узнать в соответствующей инструкции.
В этом разделе будут использоваться файлы из . Скопируем директорию с примером и переместимся в неё:
cp -r /opt/software/deepmd-kit/v2/examples/methane ~ cd ~/methaneСоздадим файл
topol.tpr с помощью команды gmx_mpi grompp, предварительно загрузив модуль для GROMACS: module purge module load GROMACS/2022.2 gmx_mpi grompp -f md.mdp -c lig_solv.gro -p topol.top -o topol.tpr -maxwarn 5 cp ~/CH4/01.train/graph-compress.pb ./Сформируем sbatch-файл gromacs.sbatch для запуска задачи на суперкомпьютере. Перед командой запуска добавим строку, которая будет указывать GROMACS использовать нашу модель:
export GMX_DEEPMD_INPUT_JSON=input.json
В файле input.json необходимо изменить значение graph_file: {
"graph_file": "graph-compress.pb",
"type_file": "type.raw",
"index_file": "index.raw",
"lambda": 1.0,
"pbc": false
}
- graph_file: файл графа (с расширением .pb), созданный командой dp freeze
- type_file: файл, в котором указаны типы атомов DP - index_file: файл, содержащий индексы атомов DP, которые должны соответствовать порядку индексов в файле .gro, начиная с нуля
Отправляем файл gromacs.sbatch на выполнение командой
sbatch gromacs.sbatch. Дополнительная информация
- Учебные материалы по DeePMD-kit
- Репозиторий на GitHub
- Инструкция для LAMMPS
- Инструкция для GROMACS
- Инструкция по работе с системой мониторинга эффективности HPC TaskMaster
![]()
Нашли опечатку?
Выделите её, нажмите Ctrl+Enter и отправьте нам уведомление. Спасибо за участие!
Сервис предназначен только для отправки сообщений об орфографических и пунктуационных ошибках.