Gromacs
👉Вернуться к списку инструкций
 Gromacs - пакет программ для моделирования физико-химических процессов в молекулярной динамике. Разработан командой Германа Берендсена, работающей в отделении биофизической химии университета Гронингена. В настоящее время развивается и поддерживается усилиями энтузиастов, в число которых входят представители университета Уппсалы и королевского технологического института. Пакет предназначен для моделирования биомолекул (например, молекул белков и липидов), имеющих много связанных взаимодействий между атомами. Обеспечивает высокую скорость расчётов для несвязанных взаимодействий.
Gromacs - пакет программ для моделирования физико-химических процессов в молекулярной динамике. Разработан командой Германа Берендсена, работающей в отделении биофизической химии университета Гронингена. В настоящее время развивается и поддерживается усилиями энтузиастов, в число которых входят представители университета Уппсалы и королевского технологического института. Пакет предназначен для моделирования биомолекул (например, молекул белков и липидов), имеющих много связанных взаимодействий между атомами. Обеспечивает высокую скорость расчётов для несвязанных взаимодействий.
Официальный сайт: http://www.gromacs.org/
Доступные версии на суперкомпьютере НИУ ВШЭ:
# Версии для очереди rocky
module load el9 # Сначала подключаем модули rocky либо заходим на login-02module load gromacs/2026_2 # Универсальная сборка Gromacs+Plumed для любых типов узлов
module loadgromacs/2026_2_intel # Сборка Gromacs+Plumed с AVX512 для узлов с CPU Intel (кроме type_e)
# Legacy-версии для очереди normal
module load GROMACS/2022.2
module load GROMACS/2020.6_plumed_v2.7.2 # С плагином Plumed 2.7.2.
module load GROMACS/2019.6
Обновление версий производится по запросу пользователей.
Тестовые исходные данные и sbatch-скрипты расположены на суперкомпьютере в каталоге /opt/software/gromacs/examples
Пользователи: Международная лаборатория суперкомпьютерного атомистического моделирования и многомасштабного анализа, Департамент прикладной математики МИЭМ, любые подразделения НИУ ВШЭ.
Выполнение расчётов на суперкомпьютере
GROMACS поддерживает параллелизм как внутри одного узла с использованием нитей, так и на нескольких узлах с использованием MPI, а также поддерживается ускорение вычисленией на GPU. На суперкомпьютере НИУ ВШЭ GROMACS скомпилирован с поддержкой Intel MPI, CUDA и OpenMP. Соответствующие модули автоматически подключаются при использовании модуля GROMACS.
GROMACS на суперкомпьютере скомпилирован с использованием смешанной точности. Вычисления, не требующие высокой точности, выполняются в одинарной точности (FP32) для повышения скорости, а критически важные этапы выполняются в двойной точности (FP64) для обеспечения корректности результатов.
Для выполнения расчёта на суперкомпьютере необходимо подготовить скрипт-файл для очереди задач с запросом требуемых ресурсов (см. инструкцию по запуску задач на суперкомпьютере).
Использование GPU-ускорителей (рекомендуется)
Запуск версии для очереди Rocky
Для тестового запуска создайте отдельную директорию и перейдите в нее:
cd ~
mkdir gmx_test
cd gmx_test
Создайте файл topol.top:
#include "oplsaa.ff/forcefield.itp"
#include "oplsaa.ff/spc.itp"
[ system ]
Water benchmark
[ molecules ]
Создайте файл em.mdp:
integrator = steep
nsteps = 2000
emtol = 1000
cutoff-scheme = Verlet
coulombtype = PME
rcoulomb = 1.0
rvdw = 1.0
Создайте файл grompp.mdp:
integrator=md
nsteps=1000000
dt=0.002
nstlist=100
cutoff-scheme=Verlet
coulombtype=PME
rcoulomb=1.0
rvdw=1.0
tcoupl=v-rescale
tc-grps=System
tau-t=0.1
ref-t=300
constraints=h-bonds
nstlog=100
nstenergy=100
В данном примере, для расчёта будет выделено 8 ядер CPU и 1 GPU на одном вычислительном узле. Содержимое файла gromacs-gpu.sbatch:
#!/bin/bash
#SBATCH --job-name=gmx-run # Название задачи
#SBATCH --partition=rocky # Очередь
#SBATCH --constraint="type_a|type_b|type_c" # Предпочтительные типы вычислительных узлов
#SBATCH --nodes=1 # Количество узлов
#SBATCH --ntasks-per-node=1 # Количество MPI процессов
#SBATCH --cpus-per-task=8 # Количество ядер CPU
#SBATCH --gpus-per-node=1 # Количество GPU
#SBATCH --time=1:00:00 # Ограничение времени
#SBATCH --output=%x-%j.out # Выходной лог-файл
#SBATCH --mail-user=ваша_почта # Укажите ваш email для отправки уведомлений (если нужно)
#SBATCH --mail-type=END,FAIL # События, требующие уведомления
# Загрузка модуля Gromacs
module purge
module load el9
module load gromacs/2026_2
# Подготовка переменных окружения
export OMP_NUM_THREADS="${SLURM_CPUS_PER_TASK:-4}"
NRANKS="${SLURM_NTASKS:-1}"
PME_OPT=""
[ "${NRANKS}" -ge 2 ] && PME_OPT="-npme 1"
# Подготовка входных файлов для моделирования
gmx_mpi solvate -cs spc216.gro -box 10 10 10 -o conf.gro -p topol.top
gmx_mpi grompp -f em.mdp -c conf.gro -p topol.top -o em.tpr -maxwarn 2
gmx_mpi mdrun -deffnm em -nb gpu -ntomp "${OMP_NUM_THREADS}"
gmx_mpi grompp -f grompp.mdp -c em.gro -p topol.top -o md.tpr -maxwarn 2
# Команда запуска
srun --mpi=pmix gmx_mpi mdrun -s md.tpr -deffnm md -nb gpu -pme gpu -bonded auto ${PME_OPT} -ntomp "${OMP_NUM_THREADS}" -pin off -maxh 0.8 -cpo md.cpt -cpt 15
# Рекомендуемые пропорции ресурсов на cHARISMa:
# -на узлах типов A, B: 1 GPU V100 и 8 ядер CPU,
# 2 GPU V100 и 16 ядер CPU.
# Как можно больше заполняйте память GPU. Отслеживайте загрузку памяти через: https://lk.hpc.hse.ru
Запуск legacy-версии для очереди Normal
В данном примере, для расчёта будет выделено 20 ядер и 1 GPU на одном вычислительном узле.
Содержимое файла gromacs-gpu.sbatch:
#!/bin/bash #SBATCH --job-name=gromacs # Название задачи # #SBATCH --partition=rocky # для новых версий подходит очередь rocky, для старых - normal
#SBATCH --time=01:00:00 # Максимальное время выполнения #SBATCH --ntasks 20 # Количество MPI процессов #SBATCH --nodes 1 # Все CPU и GPU будут на одном вычислительном узле #SBATCH --gpus 1 # Требуемое кол-во GPU
#SBATCH --constraint="type_a|type_b" # Предпочтительные типы вычислительных узлов
#SBATCH --mail-user=ваша_почта # Укажите ваш email для отправки уведомлений
#SBATCH --mail-type=END,FAIL # События, требующие уведомления
module purge # Выгрузка всех модулей
module load GROMACS/2022.2 # Загрузка строго модуля GROMACS для legacy-очереди normal
srun --mpi=pmix_v3 gmx_mpi mdrun -ntomp 1 -v -s benchmark.tpr
# Рекомендуемые пропорции ресурсов на cHARISMa:
# -на узлах типов A, B: 1 GPU и 20 ядер, 2 GPU и 40 ядер; 4 GPU и 44/48 ядер.
# Как можно больше заполняйте память GPU. Отслеживайте загрузку памяти через: https://lk.hpc.hse.ru
Для постановки задачи в очередь выполните команду sbatch gromacs-gpu.sbatch
Не забудьте скорректировать параметры в скрипте для своих расчетов!
Посмотреть состояние своих задач можно с помощью команды mj
Сообщения, выводимые в процессе работы GROMACS, будут записаны в файл gromacs-00000.err (где 00000 - номер задачи в очереди). Результат выполнения расчёта будет сохранён в файл md.log, в случае, если такой файл уже существует в текущем каталоге, он будет переименован в #md.log.1#.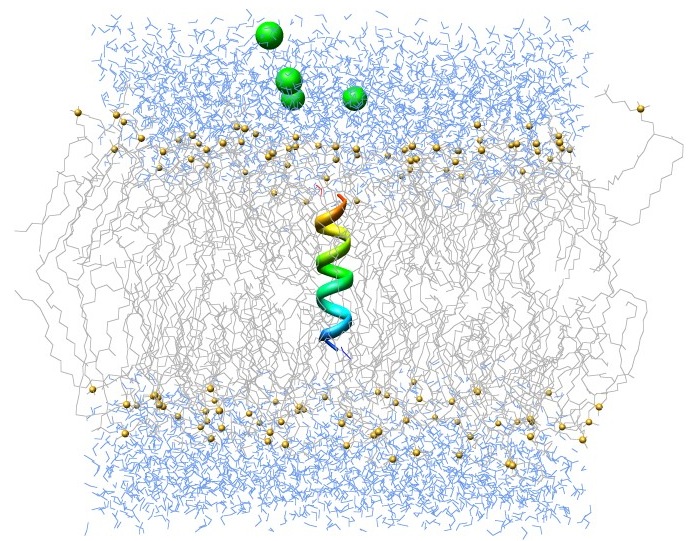 Пример симуляции мембранного белка (см. http://www.mdtutorials.com/gmx/membrane_protein/index.html).
Пример симуляции мембранного белка (см. http://www.mdtutorials.com/gmx/membrane_protein/index.html).
Запуск на CPU
Если все GPU заняты, Вы можете запускать задачу на CPU. Рекомендуемое количество ядер CPU: 24. Задача, запущенная на 24 CPU без ускориеля, будет выполняться примерно в 5 раз дольше, чем на 24 CPU + 1 GPU V100, однако, для коротких и учебных задач быстрый запуск может быть важнее увеличения времени выполнения.
Запуск версии для очереди Rocky
В данном примере, для расчёта будет выделено 16 процессорных ядер на одном вычислительном узле. Предварительно необходимо создать файлы, указанные в случае для запуска на GPU (topol.top, em.mdp, grompp.mdp).
#!/bin/bash
#SBATCH --job-name=gmx-run # Название задачи
#SBATCH --partition=rocky # Очередь
#SBATCH --constraint="type_a|type_b|type_c" # Предпочтительные типы вычислительных узлов
#SBATCH --nodes=1 # Количество узлов
#SBATCH --ntasks-per-node=1 # Количество MPI процессов
#SBATCH --cpus-per-task=16 # Количество CPU-ядер
#SBATCH --time=1:00:00 # Ограничение времени
#SBATCH --output=%x-%j.out # Выходной лог-файл
#SBATCH --mail-user=ваша_почта # Укажите ваш email для отправки уведомлений
#SBATCH --mail-type=END,FAIL # События, требующие уведомления
# Загрузка модуля Gromacs
module purge
module load el9
module load gromacs/2026_2
# Подготовка переменных окружения
export OMP_NUM_THREADS="${SLURM_CPUS_PER_TASK:-4}"
NRANKS="${SLURM_NTASKS:-1}"
PME_OPT=""
[ "${NRANKS}" -ge 2 ] && PME_OPT="-npme 1"
# Подготовка входных файлов для моделирования
gmx_mpi solvate -cs spc216.gro -box 10 10 10 -o conf.gro -p topol.top
gmx_mpi grompp -f em.mdp -c conf.gro -p topol.top -o em.tpr -maxwarn 2
gmx_mpi mdrun -deffnm em -nb cpu -ntomp "${OMP_NUM_THREADS}"
gmx_mpi grompp -f grompp.mdp -c em.gro -p topol.top -o md.tpr -maxwarn 2
# Команда запуска
srun --mpi=pmix gmx_mpi mdrun -s md.tpr -deffnm md -nb cpu -pme cpu -bonded cpu ${PME_OPT} -ntomp "${OMP_NUM_THREADS}" -pin off -maxh 0.8 -cpo md.cpt -cpt 15
Запуск legacy-версии для очереди Normal
В данном примере, для расчёта будет выделено 16 процессорных ядра на одном вычислительном узле.
Содержимое файла gromacs-cpu.sbatch:
#!/bin/bash #SBATCH --job-name=gromacs-cpu # Название задачи #SBATCH --time=24:00:00 # Максимальное время выполнения
#SBATCH --nodes 1 # Все ядра CPU будут на одном вычислительном узле
#SBATCH --ntasks 16 # Количество MPI процессов = ядер CPU
#SBATCH --mail-user=ваша_почта # Укажите ваш email для отправки уведомлений
#SBATCH --mail-type=END,FAIL # События, требующие уведомления
module load GROMACS/2022.2 # Загрузка модуля GROMACS
srun --mpi=pmix_v3 gmx_mpi mdrun -ntomp 1 -v -s benchmark.tpr
# Рекомендуемое количество ядер CPU: 24.
# Допускается запуск без указания типа узлов.
Для постановки задачи в очередь выполните команду sbatch gromacs-cpu.sbatch
Не забудьте скорректировать параметры в скрипте для своих расчетов!
Посмотреть состояние своих задач можно с помощью команды mj
Сообщения, выводимые в процессе работы GROMACS, будут записаны в файл gromacs-00000.err (где 00000 - номер задачи в очереди). Результат выполнения расчёта будет сохранён в файл md.log, в случае, если такой файл уже существует в текущем каталоге, он будет переименован в #md.log.1#.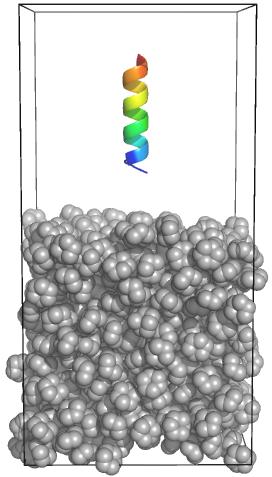 Пример построения двухфазной системы (см. http://www.mdtutorials.com/gmx/biphasic/index.html).
Пример построения двухфазной системы (см. http://www.mdtutorials.com/gmx/biphasic/index.html).
Оптимизация производительности
В GROMACS встроен модуль Wallcycle, который используется для подсчёта различных параметров производительности во время выполнения задачи. В конце лог-файла после окончания расчёта размещается раздел "Real cycle and time accounting", представляющий собой таблицу со статистикой выполнения различных частей задачи. Данная статистика может быть использована для увеличения производительности расчётов. Рекомендуется перед большими расчётами выполнять тестовые запуски с небольшим количеством шагов, чтобы подобрать наиболее подходящие параметры расчёта.
Пример такой таблицы (расшифровка названий параметров представлена в документации GROMACS):
R E A L C Y C L E A N D T I M E A C C O U N T I N G
On 24 MPI ranks
Computing: Num Num Call Wall time Giga-Cycles
Ranks Threads Count (s) total sum %
-----------------------------------------------------------------------------
Domain decomp. 24 1 100 13.124 659.868 2.7
DD comm. load 24 1 68 0.212 10.639 0.0
DD comm. bounds 24 1 42 0.014 0.718 0.0
Neighbor search 24 1 101 7.464 375.275 1.5
Launch GPU ops. 24 1 20002 1.472 74.037 0.3
Comm. coord. 24 1 9900 13.897 698.731 2.8
Force 24 1 10001 7.419 373.006 1.5
Wait + Comm. F 24 1 10001 17.554 882.618 3.6
PME mesh 24 1 10001 308.036 15488.039 63.1
Wait Bonded GPU 24 1 101 0.001 0.025 0.0
Wait GPU NB nonloc. 24 1 10001 10.014 503.505 2.1
Wait GPU NB local 24 1 10001 0.352 17.675 0.1
NB X/F buffer ops. 24 1 39802 17.784 894.201 3.6
Write traj. 24 1 3 0.527 26.488 0.1
Update 24 1 10001 10.051 505.366 2.1
Constraints 24 1 10001 43.003 2162.193 8.8
Comm. energies 24 1 1001 10.425 524.159 2.1
Rest 26.473 1331.057 5.4
-----------------------------------------------------------------------------
Total 487.820 24527.600 100.0
-----------------------------------------------------------------------------
Breakdown of PME mesh computation
-----------------------------------------------------------------------------
PME redist. X/F 24 1 20002 38.523 1936.953 7.9
PME spread 24 1 10001 95.126 4782.919 19.5
PME gather 24 1 10001 60.742 3054.086 12.5
PME 3D-FFT 24 1 20002 79.050 3974.614 16.2
PME 3D-FFT Comm. 24 1 20002 28.722 1444.147 5.9
PME solve Elec 24 1 10001 5.805 291.850 1.2
-----------------------------------------------------------------------------
Core t (s) Wall t (s) (%)
Time: 11707.660 487.820 2400.0
(ns/day) (hour/ns)
Performance: 3.543 6.775
Finished mdrun on rank 0 Tue Aug 30 10:25:00 2022
Дополнительная информация
- Документация GROMACS
- Учебники с примерами расчётов
- Оптимизация производительности GROMACS
- Ускорение вычислений на GPU
- Инструкция по работе с системой HPC TaskMaster
![]()
Нашли опечатку?
Выделите её, нажмите Ctrl+Enter и отправьте нам уведомление. Спасибо за участие!
Сервис предназначен только для отправки сообщений об орфографических и пунктуационных ошибках.