LAMMPS
👉Вернуться к списку инструкций
 LAMMPS (Large-scale Atomic/Molecular Massively Parallel Simulator) — это открытый программный пакет для решения задач молекулярной динамики, широко применяемый в научных исследованиях и инженерных расчётах. Он предназначен для моделирования поведения атомов, молекул и частиц в различных физических системах, включая твёрдые тела, жидкости, полимеры, биологические структуры и наноматериалы. Пакет поддерживает широкий набор межатомных потенциалов и моделей взаимодействия, что обеспечивает гибкость при постановке задач различной сложности. Кроме того, LAMMPS предоставляет инструменты для задания начальных условий, управления параметрами моделирования и анализа полученных результатов.
LAMMPS (Large-scale Atomic/Molecular Massively Parallel Simulator) — это открытый программный пакет для решения задач молекулярной динамики, широко применяемый в научных исследованиях и инженерных расчётах. Он предназначен для моделирования поведения атомов, молекул и частиц в различных физических системах, включая твёрдые тела, жидкости, полимеры, биологические структуры и наноматериалы. Пакет поддерживает широкий набор межатомных потенциалов и моделей взаимодействия, что обеспечивает гибкость при постановке задач различной сложности. Кроме того, LAMMPS предоставляет инструменты для задания начальных условий, управления параметрами моделирования и анализа полученных результатов.
Доступные версии на суперкомпьютере НИУ ВШЭ
# Версии для очереди rocky
module load el9 # Сначала подключаем модули rocky или заходим на login-02
# Lammps+CUDA, Intel (кроме type_e)
module load lammps/22Jul2025_update3-foss-2025b-no-kokkos-CUDA-12.9.1
# Lammps+KOKKOS, Intel (кроме type_e)
module load lammps/22Jul2025_update3-foss-2025b-kokkos
# Lammps+KOKKOS+CUDA, Intel (кроме type_e)
module load lammps/22Jul2025_update3-foss-2025b-kokkos-CUDA-12.9.1
# Lammps+KOKKOS+CUDA, AMD для type_e
module load lammps/22Jul2025_update3-foss-2025b-kokkos-CUDA-12.9.1-TYPE-E
# Legacy-версии для очереди normal
module load lammps/2022jun23_update1 # С поддержкой GPU
module load lammps/2022jun23_update1_cpu # Для расчетов без GPU
module load lammps/2022sep15
module load lammps/2022sep15_cpu
Обновление версий производится по запросу пользователей.
Тестовые исходные данные и sbatch-скрипты расположены на суперкомпьютере в каталогах /opt/software/lammps/bench/ и /opt/software/lammps/examples/
Пользователи: Международная лаборатория суперкомпьютерного атомистического моделирования и многомасштабного анализа, любые подразделения НИУ ВШЭ.
Установленные пакеты
LAMMPS на суперкомпьютере НИУ ВШЭ скомпилирован со следующим набором пакетов:
ASPHERE BOCS BODY BPM BROWNIAN CG-DNA CG-SDK CLASS2 COLLOID COLVARS COMPRESSCORESHELL DIELECTRIC DIFFRACTION DIPOLE DPD-BASIC DPD-MESO DPD-REACTDPD-SMOOTH DRUDE EFF ELECTRODE EXTRA-COMPUTE EXTRA-DUMP EXTRA-FIXEXTRA-MOLECULE EXTRA-PAIR FEP GPU GRANULAR INTERLAYER KOKKOS KSPACE MANYBODY MCMEAM MISC ML-IAP ML-SNAP MOFFF MOLECULE OPENMP OPT ORIENT PERI PHONON PLUGINPOEMS QEQ REACTION REAXFF REPLICA RIGID SHOCK SPH SPIN SRD TALLY UEF YAFF
Выполнение расчётов на суперкомпьютере
LAMMPS поддерживает параллелизм как внутри одного узла с использованием нитей, так и на нескольких узлах с использованием MPI, а также поддерживается ускорение вычислений на GPU. На суперкомпьютере НИУ ВШЭ LAMMPS скомпилирован с поддержкой CUDA и OpenMP. Соответствующие модули автоматически подключаются при использовании модуля LAMMPS.
LAMMPS на суперкомпьютере НИУ ВШЭ скомпилирован с пакетом GPU, который используется для работы с графическими процессорами. Этот пакет на суперкомпьютере использует смешанную точность: вычисления, не требующие высокой точности, выполняются в FP32 для повышения скорости, а критически важные этапы выполняются в FP64 для обеспечения корректности результатов.
Для выполнения расчёта на суперкомпьютере необходимо подготовить скрипт-файл для очереди задач с запросом требуемых ресурсов (см. инструкцию по запуску задач на суперкомпьютере).
Использование GPU-ускорителей (рекомендуется)
В данном примере, для расчёта будет выделено 16 ядер и 1 GPU на одном вычислительном узле. Использованный в примере файл in.lj находится по пути /opt/software/lammps/examples/COUPLE/simple/in.lj.
Содержимое файла lammps-gpu.sbatch для версии 2022:
#!/bin/bash #SBATCH --job-name=lammps # Название задачи #SBATCH --time=01:00:00 # Максимальное время выполнения #SBATCH --ntasks 16 # Количество MPI процессов = ядер CPU #SBATCH --nodes 1 # Все CPU и GPU будут на одном узле #SBATCH --gpus 1 # Требуемое кол-во GPU
#SBATCH --constraint="type_a|type_b|type_c" # Предпочтительные типы вычислительных узлов
#SBATCH --mail-user=ваша_почта # Укажите ваш email для отправки уведомлений (если нужно)
#SBATCH --mail-type=END,FAIL # События, требующие уведомления
module load lammps/2022jun23_update1 # Загрузка модуля LAMMPS
srun --mpi=pmix_v3 lmp -sf gpu -pk gpu $SLURM_GPUS -in in.lj # Команда запуска
# Рекомендуемые пропорции ресурсов:
# - на узлах типов A, B, C: 1 GPU и 16/24 ядер CPU, 1 вычислительный узел;
# Подбирайте пропорции под конкретную задачу.
# Как можно больше заполняйте память GPU. Иcпользуйте https://lk.hpc.hse.ru
Для версии 2025 скрипт будет следующим:
#!/bin/bash #SBATCH --job-name=lammps # Название задачи #SBATCH --time=01:00:00 # Максимальное время выполнения #SBATCH --ntasks 8 # Количество MPI процессов = ядер CPU #SBATCH --nodes 1 # Все CPU и GPU будут на одном узле #SBATCH --gpus 1 # Требуемое кол-во GPU
#SBATCH --constraint="type_a|type_b|type_c" # Предпочтительные типы вычислительных узлов
#SBATCH --partition=rocky # Очередь для запуска задачи
#SBATCH --mail-user=ваша_почта # Укажите ваш email для отправки уведомлений (если нужно)
#SBATCH --mail-type=END,FAIL # События, требующие уведомления
# Очистка системы модулей и загрузка модуля LAMMPS
module purge
module load el9
module load lammps/22Jul2025_update3-foss-2025b-kokkos-CUDA-12.9.1
# Команда запуска
export OMP_NUM_THREADS=1
srun --mpi=pmix_v3 lmp -k on g 1 -sf kk -in in.lj
# Рекомендуемые пропорции ресурсов:
# - на узлах типов A, B, C: 1 GPU и 8/16/24 ядер CPU, 1 вычислительный узел;
# Подбирайте пропорции под конкретную задачу.
# Как можно больше заполняйте память GPU. Иcпользуйте https://lk.hpc.hse.ru
Для постановки задачи в очередь выполните команду sbatch lammps-gpu.sbatch
Не забудьте скорректировать параметры в скрипте для своих расчетов!
Посмотреть состояние своих задач можно с помощью команды mj
Сообщения, выводимые в процессе работы LAMMPS, будут записаны в файл slurm-00000.out (где 00000 - номер задачи в очереди).
Запуск на CPU
Если все GPU заняты, Вы можете запускать задачу на CPU. Задача, запущенная без GPU ускорителя, будет выполняться в несколько раз дольше, однако, для коротких и учебных задач быстрый запуск может быть важнее увеличения времени выполнения. В данном примере, для расчёта будет выделено 24 процессорных ядра на одном вычислительном узле. Использованный в примере файл in.lj находится по пути /opt/software/lammps/examples/COUPLE/simple/in.lj.
Содержимое файла lammps-cpu.sbatch для версии 2022:
#!/bin/bash #SBATCH --job-name=lammps-cpu # Название задачи #SBATCH --time=24:00:00 # Максимальное время выполнения
#SBATCH --nodes 1 # Все ядра CPU будут выделены на одном узле
#SBATCH --ntasks 16 # Количество MPI процессов = ядер CPU
#SBATCH --constraint="type_a|type_b|type_c" # Предпочтительные типы вычислительных узлов
#SBATCH --mail-user=ваша_почта # Укажите ваш email для отправки уведомлений
#SBATCH --mail-type=END,FAIL # События, требующие уведомления
module load lammps/2022jun23_update1_cpu # Загрузка модуля LAMMPS без поддержки GPU
srun --mpi=pmix_v3 lmp -in in.lj # Команда запуска
Для версии 2025 скрипт будет следующим:
#!/bin/bash #SBATCH --job-name=lammps-cpu # Название задачи #SBATCH --time=24:00:00 # Максимальное время выполнения
#SBATCH --nodes 1 # Все ядра CPU будут выделены на одном узле
#SBATCH --ntasks 16 # Количество MPI процессов = ядер CPU
#SBATCH --constraint="type_a|type_b|type_c" # Предпочтительные типы вычислительных узлов
#SBATCH --partition=rocky # Очередь для запуска задачи
#SBATCH --mail-user=ваша_почта # Укажите ваш email для отправки уведомлений (если нужно)
#SBATCH --mail-type=END,FAIL # События, требующие уведомления
# Загрузка модуля LAMMPS без поддержки GPU
module load el9 module load lammps/22Jul2025_update3-foss-2025b-kokkos
srun -n $SLURM_NTASKS lmp -in in.lj # Команда запуска
Для постановки задачи в очередь выполните команду sbatch lammps-cpu.sbatch
Не забудьте скорректировать параметры в скрипте для своих расчетов!
Посмотреть состояние своих задач можно с помощью команды mj
Сообщения, выводимые в процессе работы LAMMPS, будут записаны в файл slurm-00000.out (где 00000 - номер задачи в очереди).
Оптимизация производительности
В LAMMPS встроен пакет KOKKOS, ускоряющий выполнение расчётов в ряде случаев. Наибольшее ускорение достигается при решении задач с большим количеством атомов на GPU ускорителях. Например, бенчмарк "Потенциал Леннарда-Джонса" с использованием различных пакетов (https://www.lammps.org/bench.html#accelerator) показывает следующие результаты: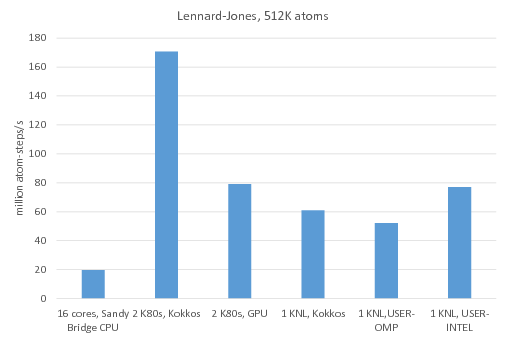
Для запуска расчёта с использованием KOKKOS изменить строку запуска. Например, для расчёта на двух GPU: lmp -k on g 2 -sf kk
Дополнительная информация по использованию KOKKOS размещается на официальном сайте LAMMPS (https://docs.lammps.org/Speed_kokkos.html).
Визуализация данных
Программа, в которой происходит визуализация - OVITO. Можно скачать с официального сайта. Небольшой пример, как работать с этой программой в видео.
Пример визуализации для этого туториала:
Дополнительная информация
- Документация LAMMPS, включая примеры расчётов и рекомендации по оптимизации
- Инструкция по работе с системой HPC TaskMaster
![]()
Нашли опечатку?
Выделите её, нажмите Ctrl+Enter и отправьте нам уведомление. Спасибо за участие!
Сервис предназначен только для отправки сообщений об орфографических и пунктуационных ошибках.